

Explaining the Ratio of Stereoisomers Produced in the Sodium Borohydride Reduction of 4-t-Butylcyclohexanone
Due to the large size of the t-butyl group, the molecule 4-t-butylcyclohexanone exists almost exclusively in one chair conformation: the one in which the t-butyl group is in the equatorial position. As a result of this conformational immobility of the molecule, the sodium borohydride reduction of 4-t-butylcyclohexanone (in ethanol solvent at 25 degrees) from one side of the molecule gives trans-4-t-butylcyclohexanol as the major product, whereas reduction from the other side of the molecule gives cis-4-t-butylcyclohexanol as the minor product (Figure 1). The identity and percent of each stereoisomer present in the product mixture are established easily by proton-NMR1.
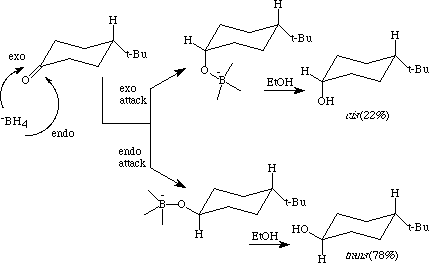
Figure 1
The question arises as to why the major stereoisomer formed is the trans isomer. In answering that question, students are likely to point to the relative thermodynamic stabilities of the two products and note that the cis isomer has greater steric strain in it (due to steric strain between the axial hydroxyl group and the axial hydrogens on carbons 3 and 5) than does the trans isomer. However, such an answer is not correct, since the reaction is irreversible, resulting in the ratio of stereoisomeric products being determined by their relative rates of formation; not by their relative thermodynamic stabilities. Thus, one must examine why the rate of attack of -BH4 from the endo side of 4-t-butylcyclohexanone (giving the trans isomer) is greater than the rate of attack of -BH4 from the exo side.
A clue to the answer can be found by making an isodensity surface of the 4-t-butylcyclohexanone molecule and coloring it with the LUMO of the molecule. Using the CAChe Editor, construct the 4-t-butylcyclohexanone molecule. From the Select menu choose Select All, and from the Beautify menu choose Comprehensive. Save the structure as 4-t-butylcyclohexanone, and then from the Applications menu select MOPAC and optimize the geometry of the 4-t-butylcyclohexanone molecule using the PM3 parameter set. Following the optimization enter the Applications menu again and select the Tabulator. In the first menu box evaluate the isosurface for the menu item electron density (not for molecular orbitals). In the second set of menu boxes make these selections: all occupied orbitals, color by LUMO, and an isosurface value of 0.010 e/A3. In the Grid Intervals menu box the default values of 41 and 0.300 Angstroms can be used. Select RUN, and when the run is complete enter the Applications menu and select the Visualizer. Open the File menu and select the 4-t-butylcyclohexanone.LonD file and open it. The resulting picture of the molecule shows an isodensity surface that displays reactive sites based upon the distribution of electrons in the lowest unoccupied molecular orbital. Regions in the molecule where there is little electron density and where the molecule is susceptible to attack by nucleophiles are shown as bright bull's eye patterns of color. As expected, for 4-t-butylcyclohexanone the bull's eye is on the carbonyl carbon. But more importantly, when the exo side of the molecule is viewed it appears that the axial hydrogens on C-2 and C-6 would offer steric hindrance to attack by the -BH4 nucleophile on the carbonyl carbon.
(Figure 2).
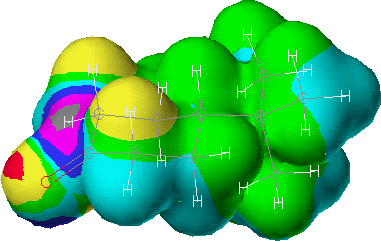
Figure 2: The exo side of 4-t-butylcyclohexanone
By contrast, when the molecule is rotated so that the endo side is viewed, then it appears that neither the equatorial hydrogens on C-2 and C-6, nor the axial hydrogens at C-3 and C-5 offer any significant steric hindrance to attack on the carbonyl carbon. (Figure 3).
Figure 3: The endo side of 4-t-butylcyclohexanone
Hence, one predicts that nucleophilic attack by -BH4 should occur predominantly from the endo side of 4-t-butylcyclohexanone, resulting in the trans stereoisomer being the major product, as is observed experimentally.
The prediction reached from a visual inspection of the two sides of the 4-t-butylcyclohexanone molecule could be put on a more quantitative foundation if we could calculate the difference in the enthalpies of activation for attack of -BH4 from the endo side versus the exo side. It is possible to do this using the CAChe system. What one does (both for exo and endo attack) is make a structure that one presumes is close to the structure of the transition state for attack of -BH4 on the carbonyl group. One considers that structure "the reactant." Then construct the structure that results from borohydride attack on the carbonyl compound, and consider that "the product." After using a program that optimizes the structures of both "the reactant" and "the product" and calculates their heats of formation, use a program that finds both the structure and the enthalpy of formation of the transition state for the reaction between "the reactant" and "the product." The difference in the enthalpy of formation of the transition state and the enthalpy of formation of "the reactant" is the enthalpy of activation for the reaction between "the reactant" and "the product."
Here are the details of the process. Using the Editor draw the 4-t-butylcyclohexanone molecule. Then draw a -BH4 on the exo face with the B atom over the O atom and one hydride H over the carbonyl carbon (Figure 4).
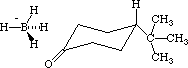
Figure 4: -BH4 poised for attack on the exo side
From the Select menu choose Select all, and from the Beautify menu choose Comprehensive. With the Select tool select the hydride H that is above the carbonyl C by clicking on it, and then (with the Shift key pressed down) select the carbonyl carbon as well. In the Adjust menu select Atom Distance, and the distance between the selected atoms will be displayed. By typing 1.600 set the H-C distance at 1.600 Angstroms (which is about 0.5 Angstroms longer than an H-C bond2 ). Click on the Apply bar and then on the Done bar. Lock the distance between those two atoms at the value by selecting Lock from the Adjust menu. In a like manner select the B and O atoms, and set the distance between them at 2.100 Angstroms (which is about 0.5 Angstroms longer than a B-O bond2 ), but do not lock the distance between those atoms. Select again the H and C that you previously locked at 1.600 Angstroms and from the Adjust menu select Unlock. Select the B, O, and carbonyl C atoms. From the Adjust menu select the Bond Angle option and enter the value of 90 degrees. Click on the Apply bar and then on the Done bar. In the same manner, set the B, H, carbonyl C bond angle to 90 degrees. Now select the B, H, carbonyl C, and O atoms. From the Adjust menu select the Dihedral Angle option and enter the value of zero. Use the Atom Tool to make single bonds between the carbonyl C and the hydride H across from it, and then between the B and O atoms. Using the Select Tool (and with the Apple key pressed down) click in the middle of the B-O, B-H, H-C, and C-O bonds, and select the menu item Weak in order to make those bonds partial bonds. From the Edit menu select Save and save this structure as the "exo reactant." From the Applications menu select MOPAC, and optimize the geometry using AM 1 parameters (MOPAC does not contain PM 3 parameters for B, so PM 3 parameters cannot be used). When the optimization is finished the DHf for "exo reactant" will be displayed, and it may have a value like -94.1 kcal/mol. Now use the Editor to build and Beautify the "exo product" as shown in Figure 5.
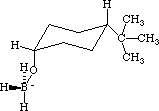
Figure 5: The exo product
Using the File menu, save the "exo product" structure. From the Applications menu select MOPAC, and optimize the geometry using AM1 parameters. The DHf for the "exo product" may have a value like -122.9 kcal/mol.
Now, in a like manner to that above, build and save both the "endo reactant" and the "endo product" that arise from nucleophilic attack of -BH4 on the endo side of 4-t-butylcyclohexanone. Likewise, use MOPAC to optimize their structures and obtain heats of formation like these: -90.5 kcal/mol ("endo reactant") and -124.1 kcal/mol ("endo product").
One is now in a position find the transition state structure and calculate its heat of formation. Form the File menu open the "exo reactant" file. From the Applications menu select MOPAC. In the menu item entitled Calculation Type, select Find Transition State. In the Parameters menu select AM1, and click on the Run bar. You will then be prompted to select the product for the calculation. Click on the OK bar, and then open the file for the "exo product". Immediately the calculation of the transition state structure and its heat of formation will begin.
A value for DHf(exo) of -73.4 kcal/mol may be obtained. This will permit calculation of the enthalpy of activation (DHact) for attack from the exo side:
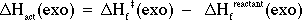
= - (73.4 kcal/mol) - (-94.1 kcal/mol)
= + 20.7 kcal/mol
In a like manner, one can obtain the enthalpy of formation for the transition state resulting from endo attack by -BH4. A value for DHf(endo) of -71.6 kcal/mol may be obtained. Therefore,
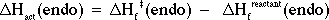
= - (71.6 kcal/mol) - (-90.5 kcal/mol)
= + 18.9 kcal/mol
Hence, the transition state resulting from -BH4 attack from the endo side of 4-t-butylcyclohexanone has the lower enthalpy of activation (by 1.8 kcal/mol) and would lead to the major product being the trans stereoisomer, as is observed. Furthermore, the value calculated (1.8 kcal/mol) for the difference between the two enthalpies of activation is fairly close to the value (0.76 kcal/mol) one calculates for the difference between the two energies of activation.
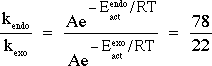
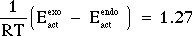
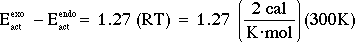
= 0.76 kcal/mol
References
- D.W. Mayo, R.M. Pike, P.K. Trumper, Microscale Organic Laboratory, John Wiley & Sons, Inc., Third Edition, 1994, p.166.
- D.D. Ebbing, General Chemistry, Houghton Mifflin Company, Fifth Edition, 1996, p.373.