

Cis-Trans Isomerism of Vision Pigments
Robert Silberman
The rod cells of the eye contain two isomers of the rhodopsin that are light sensitive. They differ in cis-trans configuration about the C11-C12 double bond. When light strikes the rod cell isomerization about the C11-C12 bond in rhodopsin take place. The isomerism results in a dramatic change in shape as the rhodopsin is converted to meta-rhodopsin.
If the molecules are light detectors, the prediction can be made that meta- rhodopsin must have a different absorption spectra than rhodopsin. It is difficult to isolate these molecules, but molecular modeling calculations can be used to determine the spectra of these two compounds. Although rhodopsins are made up of alkenes attached to the protein opsin, the spectra of the methyl analog can be used to examine the spectral properties of rhodopsin. The exercise consists of drawing the model compounds, determining their optimal conformation, determining the conformation energy of each molecule, generating electronic spectra for each molecule and comparing the two spectra. The conformation energies will be used to deduce some properties of the progress of reaction diagram.
The energy data was obtained using Mopac and AM1 from CAChe. The compounds are rhodospsin and meta-rhodopsin with a methyl group substituted for the protein portion of the molecule.
Energy Data
| compound methyl substituted | optimized structure comformation energy | excited singlet transition state energy |
| Rhodopsin | 45 kcal/mole | 194 kcal/mole |
| meta-Rhodopsin | 9.3 kcal/mole | 132.9 kcal/mole |
Electronic spectra of methyl substituted meta Rhodopsin:
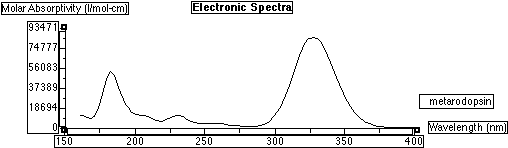
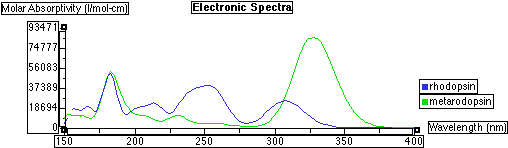
Electronic spectra of methyl substituted Rhodopsin superimposed on meta Rhodopsin: Overlay of the two spectra:
Drawing the Molecules
- Create a new folder; call it "uv-vis".
You will be saving all of your models and data to this folder, so be certain you are aware of its location on the hard drive.
- Open the CAChe Editor by choosing CAChe under the Apple menu.
If a dialog appears which asks to switch the monitor to 256 color mode, click the "OK" button.
- Draw the molecule in question.
The molecules you must draw are shown below. See the section titled "Drawing a Molecule" at the end of this document if you do not know how to draw a molecule in the CAChe Editor.
- Save the molecule. Select Mechanics from the Application menu.
- Select Optimize Structure as the calculation type. Click the "Run" button:
- When the calculation has completed, select MOPAC from the Application menu. Select Optimize Geometry as the calculation type and click the "Run" button:
- When the MOPAC calculation is complete, choose ZINDO from the Application menu.
The ZINDO application is used to construct the predicted absorption spectrum for a molecule.
- Choose Configuration Interaction (Spectra) as the calculation type and click the "Run" button:
- Once ZINDO has finished, choose Visualizer+ from the Application menu.
The Visualizer+ application is used to view the spectral data which ZINDO generates for each molecule.
- Choose Electronic Spectra from the Analyze menu.
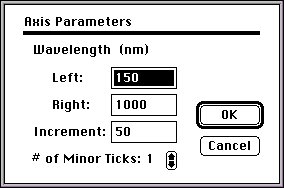
- The region of the spectrum that you will examine for lmax values is 250 nm to 350 nm. To set this range, double-click the horizontal axis of the spectrum
- Fill in the appropriate values in the dialog box and click the "OK" button:
- Record the highest point which is visible on the absorption curve. This will be your lmax value.
Repeat steps 3-13 for each of the molecules.
Drawing a Molecule
- Launch CAChe from the Apple menu. You can maximize the window size by clicking the top right box of the window.
- Select the Atom Tool from the Tool menu.
- Select Periodic Table from the Atom menu.
- Select the appropriate element from the table and click the "OK" button.
Note: Draw only non-hydrogen atoms.
- Click on the workspace. The unbonded atom will appear.
- Repeat steps 3 and 4 and select the appropriate atom which is to be bonded to the unbonded atom as described above.
- Move the cursor to the unbonded atom.
- Press and hold down the mouse button. Continue to hold down the mouse button and drag the cursor to another area of the workspace.
- Release the mouse button. The bond and atoms should now be visible. Repeat this procedure as often as needed.
- Select the Select Tool from the Tool menu.
- Using the cursor, click on the workspace (not on an atom). The entire molecule will now appear highlighted.
- Select Comprehensive from the Beautify menu to show lone electron pairs and add hydrogens.