

Product Distribution in the Nitration of Toluene
Steven W. Anderson
Notes to the Instructor: This experiment is suitable for an advanced project in the full year organic laboratory or as a project in an upper level organic course. Calculations described herein were performed using Chem CAChe. SELECTED RESULTS may be provided to students for guidance in performing their calculations. The Complete Data are available for comparison, and a separate page provides a Discussion of the complete results.
Introduction
Electrophilic aromatic substitution represents an important class of reactions in organic synthesis and is arguably the most critical transformation of arenes. The nitration of benzene and its derivatives has been extensively investigated. Nitration of toluene proceeds with predominant formation of the ortho isomer1 for which the results of Brown and Nelson2 are representative. (Fig. 1).
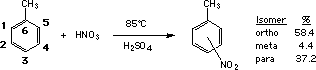
Figure 1 Nitration of toluene
The distribution of isomers does not appear to be altered significantly by changes in concentration of the nitrating agent2 but variation in reaction conditions can have a marked effect on product ratios3. Reported yields for the ortho isomer have ranged from 59 % (HNO3 in acetic anhydride4) to 69% (NO2+PF6- in nitromethane5). [The numbering scheme used on the benzene skeleton is referenced according to the carbon numbers assigned when the structure was drawn in the Editor of Chem CAChe.] This reaction is generally believed to proceed6 via an SEAr mechanism (Fig. 2).
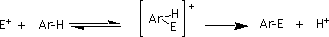
Figure 2 General Scheme for the SEAr Reaction
You have learned that ortho and para attack of the nitronium ion is favored due to stabilization of specific ortho and para resonance structures via an electron-donating inductive effect of the methyl group. The regioselectivity of the final product distribution should reflect the relative stabilities of the corresponding arenium ions in accord with the Hammond postulate8.
Energy calculations for the starting material, intermediate arenium ions, and products in the nitration of toluene can be compared with the experimental data. Heats of reaction are easily inferred from these data.
ChemCAChe, and Project Leader also permit calculation of electrophilic and nucleophilic susceptibility9 for the ring carbons of toluene and the arenium ions. The electrophilic frontier density is a measure of the susceptibility of the substrate to attack by an electrophile. Electrophilic frontier density reveals reactive sites based on the electron distribution of active orbitals near the HOMO. On the other hand, the nucleophilic frontier density is a measure of the susceptibility of the substrate to attack by a nucleophile. This parameter reveals reactive sites based on the electron distribution of active orbitals near the LUMO.
Nucleophilic susceptibility may be applied to the arenium ions generated from ortho, meta or para attack of toluene by the nitronium ion. While the arenium ions are not attacked by a nucleophile per se, charge distribution might be inferred from the resultant data. Values for carbons bearing a positive charge, through resonance, indicate the degree of positive charge with the larger number reflecting a greater charge.
Finally, electron isodensity surfaces may be calculated for toluene and the various arenium ions to visually determine the most likely site for electrophilic attack on toluene and the most likely site for nucleophilic attack (i.e., the most electron deficient region) on a given arenium ion.
Objectives of this Experiment
- Calculate:
- energies (steric, total, heat of formation) for the starting material, intermediate arenium ions, and products in the nitration of toluene.
- heats of reaction for starting materials to the intermediate arenium ions as well from the starting materials to the products.
- the electrophilic susceptibility for each ring carbon of toluene.
- the nucleophilic susceptibility of carbons possessing a positive charge through resonance for each of the intermediate arenium ions.
- Generate:
- an electrophilic isodensity surface for toluene.
- a nucleophilic isodensity surface for each of the arenium ions.
- Correlate all of these findings with the observed product ratios to assess the degree of agreement to experiment.
- Examine the data to see if they account for the lower than statistically expected degree of ortho product.
- Ascertain whether it can be determined if nitration, under these conditions, is governed by kinetic or thermodynamic control and whether or not the results are in agreement with the Hammond postulate.
Experimental
Note: These instructions were developed using CAChe v. 4.0 for the Macintosh but could be easily transferred to a Windows platform.
Editor Instructions
- Draw structures for toluene, the three mononitration products, and all of the intermediate arenium ions using the CAChe Editor.
- Keep molecules in the same orientations (plane of benzene ring) so the carbon numbers are retained throughout.
- Watch changes in charges and hybridization when drawing the various resonance forms for the arenium ions.
- In depicting structures, go to the VIEW menu, select "atom shape". In the "atom shape" dialog box set the following: Display atoms as: atom label consisting of -- element & charge, atom number, spheres, shaded, radius: van der Waals, scale: 0.020, polyhedra scale factor: 1.00, probability: 0.500
Project Leader Instructions
- From the Project Leader menu select a molecule or ion and analyze as follows:
- Energy Calculations: Set: Property of "chemical sample" using the property of "steric energy" and the "standard procedure". Repeat this for each sample adding the properties of "energy total" and "heat of formation".
- Susceptibility Calculations: Set: Property of "atom" using the property of "electrophilic susceptibility" for toluene and "nucleophilic susceptibility" for the arenium ions and the "standard procedure". Be careful to enter the carbon atom numbers to be evaluated ("atom number" column) manually to avoid analysis of all carbons unless this is desired. In this experiment C1-C6 of toluene were analyzed, and three carbons for the arenium ions generated by ortho/para attack (C2, C4, C6) and meta attack (C1, C3, C5). The latter were the carbons that, in the canonical resonance forms, carried positive charges. Note: these are the assigned carbon numbers from Fig. 1.
Tabulator Instructions
- Evaluate isosurfaces for: "electron density"
- Click on: "HOMO and LUMO only, color frontier density, "electrophilic" when analyzing toluene, "nucleophilic" when analyzing the arenium ions
- Set the following: grid intervals to: maximum # of 41, "preferred distance" to 0.300 Angstroms,isosurface value: 0.010 e/A3, frontier weight = 3.00
- To get meaningful color patterns, use all molecular orbitals (from HOMO -9999 to LUMO +9999) in MOPAC as well as in the Tabulator11.
- Do not save "planes" or "grids"
Visualizer Instructions
- Open the Visualizer menu
- When prompted for a molecule open the name with the ".NfonD" suffix (for nucleophilic electron density) or the ".EfonD" suffix (for electrophilic electron density).
- Once the structure is in view (type
 F for Mac platform or ctrl-F on a PC platform if it is not in view) use the track ball to scale, rotate, or translate as necessary.
F for Mac platform or ctrl-F on a PC platform if it is not in view) use the track ball to scale, rotate, or translate as necessary. - Access the VIEW pull down menu to "surface legend". This reveals a "Color Table" providing probability values for the color boundaries. Probability for both electrophilic and nucleophilic attack decreases in order of the regions colored: grey, red (0.005 at grey-red interface), yellow, green, aqua, blue, to purple and black (0 at black-purple interface). The color scheme may be set to user preferences10,11.
- Close the "Color Table" by clicking OK.
- Again access the VIEW pull down menu to "view settings". In this submenu adjust the "near clip" to get the visible depth at a contour slice close enough (generally 16-18 Angstroms seemed to work good) to gain an adequate view of the electron density. The aim is to compromise between getting too close (all carbon atoms black or parts of the ring skeleton disappear) or too far to see details of inner electron density. The "eye separation" was not active (grayed out), while the "viewing distance" and "far clip" controls were not used. The "view direction" keys (x,y,z) were also not employed.
Questions
- How does a methyl group stabilize the sigma complex?
- Are the energy calculations consistent with the known directing effect of a methyl substituent?
- Is there a correlation between charge delocalization and sigma complex stability?
- Are the energy calculations consistent with the Hammond postulate? Hint: First compare energies for sigma complex formation and then energies for the overall reaction.
- Are the values for the heats of reaction positive or negative? Explain this observation.
- Based upon your results, is the use of nucleophilic susceptibility valid in ascertaining localization of of positive charge in arenium ions? Why or why not?
Extensions and Variations
- Repeat the calculations using the COSMO solvent model in MOPAC to see how this affects calculated parameters. Explain any differences.
- Perform calculations for the nitration of t-butylbenzene. Experimental data reveal only 16% of the ortho isomer and 73% of the para isomer are formed2. Compare your findings and account for any differences.
- Other comparable programs to Chem CAChe could be employed12,13 however, the outstanding spreadsheet capability provided by Project Leader greatly expedites computation time for the multiple calculations required.
Acknowledgement
I would like to express my sincere appreciation to Doug Preston of Oxford Molecular Group, Inc. for extensive e-mail correspondence which greatly clarified the parameters used in MOPAC, ZINDO and, in particular, the Fukui electrophilic and nucleophilic electron density calculations.
References
- Stock, L.M. "A Classic Mechanism for Aromatic Nitration" in Progr. Phys. Org. Chem. 1976, 12, 21-47.
- Nelson, K.L. and Brown, H.C. J. Am. Chem. Soc. 1951, 73, 5605-5607.
- Berliner, E. Progr. Phys. Org. Chem. 1964, 2, 253.
- Ingold, C.K. "Structure and Mechanism in Organic Chemistry", 2E, Cornell University Press, Ithaca, NY, 1969, 264-417.
- Olah, G.A. Acc. Chem. Res. 1970, 4, 240-248 and references cited therein.
- Carroll, F.A. "Perspectives on Structure and Mechanism in Organic Chemistry", 1E, Brooks/Cole (1998), pp. 513-520.
- March, J. "Advanced Organic Chemistry", 4E, John Wiley, New York, 1992, pp. 511-512.
- Hammond, G.S. J. Am. Chem. Soc. 1955, 77, 334.
- Fukui, K., Yonezawa, T. , Nagata, C. and Shingu, H. J. Chem. Phys., 1953, 11, 1433-1442.
- Personal communication with Dr. Douglas Preston of Oxford Molecular Group, Inc., Beaverton, OR, 13 July 1998.
- CAChe WORKSYSTEM, CAChe Reference (Ver. 3.7, 1994), p. 3-8, CAChe Scientific, Inc. (Oxford Molecular Group, Inc.)
- Hehre, W.J., Shusterman, A. J., Huang, W. W. "A Laboratory Book of Computational Organic Chemistry", Wavefunction, Inc., Irvine, CA, 1996, Expt. 53, pp. 175-176.
- Hehre, W.J., Shusterman, A.J., Nelson, J.E. "The Molecular Modeling Workbook for Organic Chemistry", Wavefunction, Inc., Irvine, CA, 1998, Expts. 4 & 5 in Chap. 13, pp. 189-190.
- Maron, S.H., Lando, J.B. "Fundamentals of Physical Chemistry," Macmillan, New York, NY, 1974, p. 274.