

Molecular Modeling of Some Monosaccharides in Their Mutarotation Equilibria
Do Ren Chang
Introduction
In organic chemistry, we learn that open-chain alkanes differ from their counterpart cycloalkanes and no interconversions occur between them in free form. Cycloalkanes of small member rings exhibit torsional strain and angle strain, thus five- and six-membered rings are most common. The six-membered ring in the chair conformation is especially is free of angle and torsional strains. With consideration of van der Waals interactions among the ring axial and equatorial positions, the contributions of different conformers can be estimated. However, for monosaccharides, among sugars of five and six carbons atoms interconversion equilibria between open chain and rings do exist.
Because sugars are polyhydroxyaldehydes or polyhydroxyketones, intramolecular hemiacetal or hemiketal formation generates five- and six-membered rings by a process catalyzed by acids and bases. Ring closure in monosaccharides occurs by nucleophilic attack of the oxygen atom on the carbonyl carbon atom. This process creates a new chiral center responsible for the a- and b-anomers. The correspondence of open chain structures and the corresponding Haworth structures is covered in most organic texts (1).
The equilibration among the different ring sizes and anomeric configurations is accompanied by a change in optical rotation known as mutarotation. The positions of the equilibria, i.e., the compositions of equilibrated mixtures of isomeric forms of sugars solutions, can, in principle, be determined by nmr spectroscopy, and X-ray diffraction analysis of crystalline samples provides information on ring shape. An excellent review on monosaccharides can be found in chapter 2 of Collins and Ferrier’s book (2). We have applied molecular modeling to understand why in solution hemiacetal ring formation is favored for sugars like ribose, fructose, and glucose. For compounds in aqueous solution their solvation energies were considered with the hope the results would shed some light on their significance.
Consider A in equilibrium with B, C, D, and E simultaneously, as shown in the following diagram:
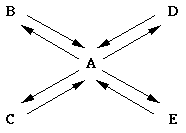
The equilibrium constants, Kab, Kac, Kad, and Kae are defined as ratios of activities of B, C, D, and E to activity of A , respectively. The equilibrium constant is related to Gibbs free energy change under standard conditions, DGo = -RTlnK and DGo = DHo - TDSo. In forming a cyclic hemiacetal from a straight-chain sugar, some degrees of freedom may have been lost and disruption of aqueous orderliness may have a similar magnitude. The conversion from carbonyl and alcohol groups into a hemiacetal changes a p bond to a s bond in an energetically favorable process:
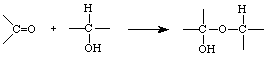
We expect that the Gibbs free energy change can be approximated by the enthalpy or energy change under these conditions. Molecular modeling software PC Spartan Pro from Wavefunction, Inc. was used for calculation of heats of formation and energies. Semi-empirical models AM1 and PM3 and a Hartree-Fock STO-3G minimal basis set and 3-21G split-valence basis set are included for comparison.Other calculations used CAChe on a Power MacIntosh G3 by selecting Mechanics from Applications menu to minimize energy, then selecting MOPAC for calculation. In Table 1, the DDH and DE refer to difference between the solvated heat of formations and Hartree-Fock energy of hemiacetals and solvated open-chain structures, respectively. Here 1 au = 627.51 kcal/mol, and s-D-sugar refers to open-chain or acyclic sugar.
Results and Discussion
| Table 1. Aqueous solvated sugar | |||||||||
PM3 |
AM1 |
STO-3G |
|||||||
| compound | DHf(au) | e-DDH/RT | % | DHf(au) | e-DH/RT | % | E(au) | e-DE/RT | % |
| a-D-glucopyranose | -0.4525 | 2.16 x 104 | 0.20 | -0.5065 | 9.11 x 1011 | 93.55 | -674.51 | 4.06 x 1032 | 49.28 |
| b-D-glucopyranose | -0.4580 | 1.08 x 105 | 1.02 | -0.5028 | 1.73 x 1010 | 1.78 | -674.51 | 1.27 x 1032 | 15.41 |
| a-D-glucofuranose | -0.4508 | 3.45 x 103 | 0.03 | -0.4993 | 4.03 x 108 | 0.04 | -674.51 | 2.70 x 1032 | 32.74 |
| b-D-glucofuranose | -0.4583 | 1.05 x 107 | 98.75 | -0.5037 | 4.51 x 1010 | 4.63 | -674.50 | 2.12 x 1031 | 2.57 |
| s-D-glucose | -0.4431 | 1 | 0.00 | -0.4806 | 1 | 0 | -674.44 | 1 | 0 |
| a-D-ribopyranose | -0.3819 | 3.09 x 104 | 5.96 x 10-7 | -0.4250 | 6.97 x 1012 | 98.79 | -562.09 | 2.22 x 1030 | 72.46 |
| b-D-ribopyranose | -0.3797 | 2.92 x 103 | 5.62 x 10-8 | -0.4192 | 1.50 x 1010 | 0.21 | -562.09 | 3.78 x 1027 | 12.34 |
| a-D-ribofuranose | -0.3835 | 1.70 x 105 | 3.28 x 10-6 | -0.4200 | 3.41 x 1010 | 0.48 | -562.09 | 2.93 x 1029 | 9.58 |
| b-D-ribofuranose | -0.3998 | 5.19 x 1012 | 100 | -0.4201 | 3.66 x 1010 | 0.52 | -562.09 | 1.72 x 1029 | 5.62 |
| s-D-ribose | -0.3722 | 1 | 0 | -0.3971 | 1 | 0 | 1 | 0 | |
| a-D-fructopyranose | -0.4566 | 1.28 | 1.9 | -0.5006 | 1.71 x 106 | 1.14 | -674.51 | 2.79 x 1030 | 0.35 |
| b-D-fructopyranose | -0.4506 | 0.0022 | 3.34 x 10-3 | -0.4931 | 582 | 3.89 x 10-4 | -674.50 | 1.50 x 1027 | 0 |
| a-D-fructofuranose | -0.4603 | 64.73 | 96.59 | -0.4956 | 8.47 x 103 | 0.00566 | -674.50 | 1.69 x 1029 | 0.02 |
| b-D-fructofuranose | -0.4519 | 0.00926 | 0.0138 | -0.5049 | 1.48 x 108 | 98.85 | -674.51 | 8.04 x 1032 | 99.63 |
| s-D-fructose | -0.4563 | 1 | 1.49 | -0.4871 | 1 | 0 | -674.44 | 1 | 0 |
HF-321G |
MOPAC |
expt.% |
|||||||
| E(au) | e-DE/RT | % | Hf(kcal/mol) | e-DHf/RT | % | Ref.3 | Ref.4 | ||
| a-D-glucopyranose | -679.562 | 3.99 x 1011 | 99.70 | -325.0040 | 2.57 x 109 | 99.38 | 37.3 | 38 | |
| b-D-glucopyranose | -679.557 | 1.20 x 109 | 0.30 | -321.9950 | 1.60 x 107 | 0.62 | 62.6 | 62 | |
| a-D glucofuranose | -679.546 | 1.66 x 104 | 4.15 x 10-6 | -273.5680 | 4.75 x 10-29 | 0 | - | - | |
| b-D-glucofuranose | -679.544 | 1.13 x 103 | 2.82 x 10-7 | -275.7200 | 1.80 x 10-27 | 0 | 0.1 | 0.14 | |
| s-D-glucose | -679.537 | 1 | 0 | -312.1760 | 1 | 0.002 | 0.02 | ||
| a-D-ribopyranose | -566.300 | 9.56 x 1012 | 12.65 | -268.8210 | 9.90 x 104 | 57 | 21.5 | 20 | |
| b-D-ribopyranose | -566.302 | 6.60 x 1013 | 87.35 | -265.5820 | 417 | 0.24 | 58.5 | 56 | |
| a-D-ribofuranose | -566.283 | 1.24 x 105 | 1.64 x 10-7 | -266.9830 | 4.44 x 103 | 2.55 | 6.5 | 6 | |
| b-D-ribofuranose | -566.289 | 3.66 x 107 | 4.84 x 10-5 | -268.6150 | 6.99 x 104 | 40.23 | 13.5 | 18 | |
| s-D-ribose | -566.272 | 1 | 0 | -262.0110 | 1 | 0 | 0.05 | ||
| a-D-fructopyranose | -679.561 | 1.55 x 1015 | 95.4 | 2.5 | |||||
| b-D-fructopyranose | -679.558 | 7.36 x 1013 | 4.54 | 65 | |||||
| a-D-fructofuranose | -679.545 | 7.05 x 107 | 4.35 x 10-6 | 6.5 | |||||
| b-D-fructofuranose | -679.554 | 9.96 x 1011 | 0.0615 | 25 | |||||
| s-D-fructose | -679.528 | 1 | 0 | 0.8 | |||||
All calculations show that cyclic hemiacetals are more stable than open-chain structures in aqueous solutions. The cyclic hemiacetals have lower energies of formation and Hartree-Fock energies. In general, the pyranose rings are heavily favored over furanose forms for aldoses, in line with the relative thermodynamic stabilities of six-membered rings over five-membered rings, except in PM3 which favors the furanose form. For ketoses the furanose is favored except in the HF-3-21G calculation.
The calculated equilibrium ratios are quite different from experimental results with respect to anomeric composition. The a/b anomeric ratio will be affected to some extent by steric interactions and by polarities of hydroxyl groups in axial or equatorial positions and also by temperature. In all calculations 25o C is used. In glucopyranose the b-anomer has all hydroxyl groups in equatorial positions, best fitting into water structure for stabilization. Our calculations indicate the a-anomer dominates. Thus, it appears that solvation energies of the b-anomer are underestimated in these calculations.
There are several possible reasons for the discrepancies: the majority of models used in calculations are based on molecular mechanics and quantum mechanics with a Hartree-Fock minimal basis set and semi-empirical models. Evaluating the composition ratio with the Boltzman factor involves the difference between two large numbers. The results are very sensitive to deviations. The system may require extended Hartree-Fock and density functional models calculations. The contributions of solvation energies need to be evaluated correctly. They are hard to evaluate, and entropy contributions and hydrogen bonding from solvent system need to be properly factored in. In any case, the solvation energies are listed in Table 2 along with non-solvated sugar energies for discussion.
| Table 2. Nonsolvated sugar energies and their energies of solvation | |||||||||
PM3 |
AM1 |
STO-3G |
HF 3-21G |
||||||
| Hsol | Hsol | Esol | Esol | ||||||
| Hf(au) | (kcal/mol) | Hf(au) | (kcal/mol) | E(au) | (kcal/mol) | E(au) | (kcal/mol) | ||
| a-D-glucopyranose | -0.4289 | -14.789 | -0.4845 | -13.819 | -674.4825 | -14.863 | -679.5400 | -13.8180 | |
| b-D-glucopyranose | -0.4297 | -15.236 | -0.4801 | -14.236 | -674.4806 | -15.379 | -679.5340 | -14.2360 | |
| a-D-glucofuranose | -0.4240 | -22.128 | -0.4790 | -12.714 | -674.4828 | -14.416 | -679.5260 | -12.7140 | |
| b-D-glucofuranose | -0.4274 | -19.002 | -0.4790 | -15.474 | -674.4786 | -15.520 | -679.5190 | -15.4730 | |
| s-D-glucose | 0.4145 | -17.971 | -0.4634 | -10.745 | -674.4207 | -9.174 | -679.5370 | -10.5170 | |
| a-D-ribopyranose | -0.3587 | -14.573 | -0.4075 | -10.998 | -562.0710 | -11.598 | -566.2830 | -10.9970 | |
| b-D-ribopyranose | -0.3608 | -11.862 | -0.4017 | -10.975 | -562.0699 | -11.226 | -566.2850 | -11.0810 | |
| a-D-ribofuranose | -0.3589 | -15.419 | -0.4015 | -11.596 | -562.0687 | -11.815 | -566.2640 | -12.0980 | |
| b-D-ribofuranose | -0.3625 | -23.398 | -0.3988 | -13.337 | -562.0665 | -12.897 | -566.2670 | -13.3370 | |
| s-D-ribose | -0.3463 | -16.195 | -0.3802 | -10.609 | -562.0072 | -10.234 | -566.2550 | -10.6110 | |
| a-D-fructopyranose | -0.4267 | -18.752 | -0.4801 | -12.872 | -674.4851 | -13.023 | -679.5430 | -11.1390 | |
| b-D-fructopyranose | -0.4248 | -16.197 | -0.4735 | -12.305 | -674.4782 | -12.916 | -679.5380 | -12.3050 | |
| a-D-fructofuranose | -0.4297 | -19.175 | -0.4707 | -15.646 | -674.4783 | -15.626 | -679.5200 | -15.6460 | |
| b-D-fructofuranose | -0.4237 | -17.704 | -0.4813 | -14.798 | -674.4888 | -14.048 | -679.5330 | -13.0520 | |
| s-D-fructose | -0.4215 | -21.887 | -0.4629 | -15.221 | -674.4191 | -12.985 | -679.5030 | -15.0000 | |
Literature Cited
- Vollhard, K. Peter.; Schore, .N.E. Organic Chemistry, 3rd. ed, chapter 24, published by W. H. Freeman, 1999.
- Collins, P.; Ferrier, R. Monosaccharides, chapter 2, by John Wiley & Sons, 1995.
- Dictionary of Organic Compounds, 6th. ed., vol. 4, p 3330, and vol. 6, p 5619, by Chapman and Hall, 1996.
- (a) Pigman, W., Isbell, H.S. Adv. Carbohydr. Chem. & Biochem. 1968, 23, 11. (b) Isbell, H. S., Pigman, W. Adv. Carbohydr. Chem. & Biochem. 1969, 24, 13.