

Product Distribution in the Nitration of Toluene
DISCUSSION
a) Results from the literature
Studies have supported the initial formation of a weak reagent-substrate ("encounter") complex from the association of the nitronium ion and the arene which is followed on the reaction coordinate by a sigma-complex7. Considerable experimental data of several groups is consistent with the rate-limiting formation of arenium ions1. Compelling evidence, from several fronts1, points to the energy requirements for the conversion of encounter complexes to the arenium ions as dictating product distribution. Preference for ortho substitution is observed in spite of possible steric hindrance. Pople CNDO/2 calculations by Olah , et. al demonstrated that the charge density is highest around the ortho and para positions which, when coupled with the 2:1 availability of ortho:para positions, rationalizes the observed preference5. Stock has estimated1 the energy difference at the transition state, for formation of the meta and para nitro arenium ions of toluene from the encounter complex, to be about 1.7 kcal/mol.
Purely statistical analysis, without consideration of the electron donating inductive effect of the methyl group, would predict 67% ortho and 33% para products. Olah and co-workers5 have shown that the phenonium ion arising from protonation of benzene, studied by NMR and semi-empirical molecular modeling, has the approximate charge distribution shown below in Figure 5.
Figure 5. Approximate charge
distribution in the phenonium ion
If used as a model for the arenium ion in electrophilic aromatic substitution, this would indicate that a para substituent has a greater effect on the adjacent carbon than an ortho substituent. In the absence of other effects this would predict7 a product ratio, for toluene, of greater than 33% para and less than 67% ortho substitution since the sigma complexes would be stabilized in the order para > ortho > meta. This accounts for the lower than expected ortho/para isomer ratio.
b) Calculated results from Chem CAChe
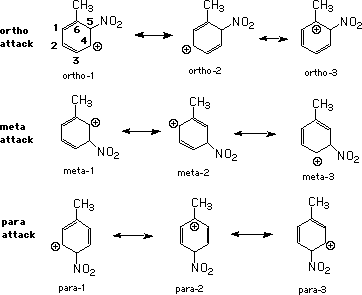
For reference, the arenium ions have been keyed as shown below.
i/ Consideration of the average energetic data for the isomers
The steric energies of each of the isomeric products are clearly in the expected order with o-nitrotoluene highest and p-nitrotoluene lowest. For the arenium ions, the average steric energies for each set are, in kcal/mol: ortho, 2.442; meta, 2.792; para, 2.438. It is not readily apparent why the ortho resonance forms would be comparable to the para forms yet lower than meta. Since this is a molecular mechanics calculation (MM3) which considers van der Waals interactions, closer inspection of the optimized structural drawings using various view representations and molecular orientations might shed light on these trends.
The total energy calculations, which consider the interaction between the electrons and the nuclei as well as the electron clouds with each other, favor the meta and para products as expected. For the arenium ions, averages are: ortho, -77.084; meta, -77.076; para, -77.096. These lean toward the ortho and para forms in agreement with laboratory results. Moreover, the para form is of lowest energy as predicted and calculated by Olah5.
The heats of formation for the products are consistent with expectations placing o-nitrotoluene highest and the para isomer lowest. The arenium ions average: ortho, 216.016; meta, 218.346; para, 213.362. These results are in complete agreement with the work of Olah5 and others1. The approx. 5 kcal/mol difference between the meta and para arenium ions compares favorably with Stock's transition state estimate.
Heats of reaction are calculated from: DHrxno = SDHfo(products) - SDHfo(reactants)14. The overall heats of reaction are moderately exothermic favoring the meta and para products perhaps reflecting the larger steric energy for the ortho isomer. The heats of reaction to the arenium ions are highly endothermic favoring the ortho and para forms, again in support of laboratory results.
The Hammond Postulate8 predicts that the kinetic product will be the one which results from the most stable arenium ion. Under these conditions (gas phase), this notion is supported owing to the lower heats of formation and lower heats of reaction for the ortho and para arenium ions. It should be noted that these calculations do not account for entropy changes which will be significant over the course of the reaction. Furthermore, the heats of formation were calculated for isolated gas phase species. It would be instructive to repeat the calculations using the COSMO solvent model in MOPAC to see how this would affect calculated parameters.
ii/ Consideration of the energetic data within given sets of resonance forms
(see Table 3 in Complete Data)
Examination of the steric energies of the resonance forms reveals that there are significant differences within each canonical set with the order. It is not readily apparent why ortho-3 ranks lowest of all of the canonical forms. Since the total energies (discussed below) are very similar, the steric differences might be revealed, as mentioned earlier, by closer inspection of the optimized structural drawings using various view representations and molecular orientations.
The total energies of the resonance forms are essentially identical within each set. Ortho-3 is the lowest for the ortho forms where the positive charge resides on C6 receiving maximum stabilization from the pendant methyl group. Meta-1 and meta-2 are of lowest energy where the positive charge is closest to the methyl group. The para forms show the least differentiation with para-3 ranking the lowest by ca. 0.6 kcal. It is not clear why para-2 is not of the lowest energy as anticipated.
The heats of formation of the resonance forms are also very close within each set. Ortho-3, where inductive stabilization would be greatest, is intermediate in energy. Meta-3, where stabilization would be least apparent, is of the lowest energy. Among the para forms, para-2 is of the lowest energy as expected.
The heats of reaction to the arenium ions are again essentially identical within each set. The same trends as for the heats of formation are observed here.
iii/ Consideration of the nucleophilic and electrophilic susceptibility data
(see Table 4 in Complete Data)
The numerical electrophilic susceptibility data for toluene indicates that the highest probability of attack is at C-3 (para) and C-6 (ipso). The meta carbons (C-2 and C-4) are least favorable. In addition to other factors, the statistical advantage of the two other carbons must outweigh the para preference. It should be noted that steric effects are included in these calculations which makes the ipso preference especially notable. This observation emphasizes the electron donating ability of a methyl group.
The electrophilic susceptibility electron density map for toluene (see Figure 3 in Selected Results) is consistent with these findings showing a preference for electrophilic attack at C-3 and C-6. However, what this map further indicates is that there is a greater region of electrophilic susceptibility in the vicinity of the ortho carbons in areas where the pi electrons would be delocalized. Nonetheless, the greatest probability centered on any carbon is at the para position. These results are entirely consistent with the data of Olah5.
Nucleophilic susceptibility data for the arenium ions may provide us with a measure of which carbon in the ring best accommodates the positive charge which, in turn, should indicate the relative contribution of a given resonance structure. The numerical data shows that all of the meta resonance forms all favor a positive charge on C1. One reason why this may be favored, in addition to other factors mentioned previously, is that this is the most sterically accessible site for nucleophilic attack. All of the para resonance forms favor C6 with the largest overall susceptibility value as would be expected with charge delocalization. The ortho forms display somewhat of a dichotomy. Ortho-1 clearly favors C2 while ortho-2 and ortho-3 slightly favor C2 over C6. Steric factors might also account for the lower than expected susceptibility value at C6 in ortho-1. If these are a valid consideration, a reasonable assumption in considering nucleophilic attack, a shadow of doubt is cast upon the use of nucleophilic susceptibility in assessing relative charge distribution in arenium ions.
The nucleophilic susceptibility electron density map for ortho-3 (see Figure 4 in Selected Results) is consistent with the numerical data mentioned above. Since the nitro group would exert a stronger inductive effect than the methyl group, it makes sense that the positive charge may be favored more on C2 as in ortho-2.